ARG1-D este o tulburare distinctă a ciclului ureei (UCD)8.
ARG1-D, cunoscută și sub denumirea de argininemie, este o boală autozomal recesivă cauzată de mutații ale genei ARG1 care codifică enzima arginază-1 (ARG1)2,9.
Enzima ARG1 descompune arginina în ornitină și uree, în ultima etapă a ciclului ureei. Ciclul ureei cuprinde cinci etape principale, are loc mai ales în ficat și transformă amoniacul (NH3), derivat din descompunerea proteinelor, în uree10.
- Arginina este un aminoacid care joacă un rol important în menținerea homeostaziei vasculare și a altor funcții fiziologice11.
- În absența unei enzime funcționale, arginina și metaboliții înrudiți cu arginina, inclusiv amoniacul și compușii derivați de la guanidină, se acumulează și sunt asociați cu patologia neuromotorie12,13.
- Nivelurile crescute persistente de arginină la pacienții cu ARG1-D sunt factori cheie ai manifestărilor bolii, incluzând spasticitatea progresivă, întârzierea dezvoltării și apariția de convulsii4,5,14,15.
- Hiperamoniemia nu este un semn distinctiv al ARG1-D și episoadele acute de hiperamoniemie apar rar14-16.
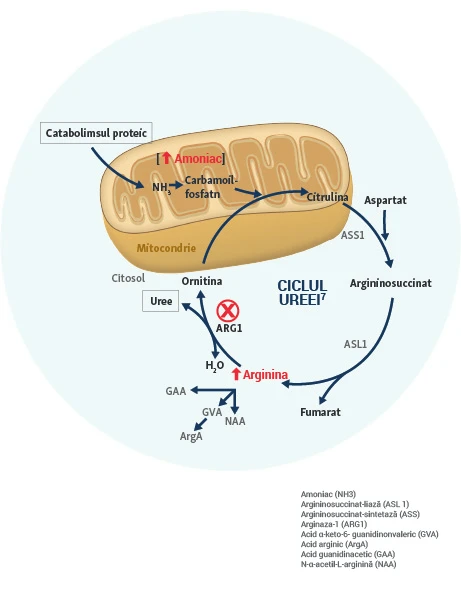
Creșterea persistentă a argininei plasmatice și a metaboliților săi îi supune pe pacienți unui risc crescut de morbiditate semnificativă și mortalitate precoce1-7.
Afectând copiii și continuând până la vârsta adultă, pacienții au manifestări heterogene ale ARG1-D2,5,18-20.
Manifestările cheie care pot fi prezente includ: spasticitate progresivă, întârziere în dezvoltare, dizabilitate intelectuală și convulsii2,5,19.
- Manifestările încep de obicei în copilărie și persistă în timp datorită acumulării nivelurilor de arginină plasmatică2.
- Spasticitatea se manifestă de obicei după primele luni de viață și se agravează progresiv până la vârsta adultă2,9,12,16.
- Diplegia spastică și mersul în vârful picioarelor sunt manifestări frecvente ale ARG1-D2,16.
- Debutul inițial este de obicei limitat la membrele inferioare; cu toate acestea, pe măsură ce spasticitatea se înrăutățește, membrele superioare pot fi și ele afectate2.
- Pacienții prezintă un declin progresiv și variabil al abilităților neurologice, de dezvoltare și funcționale2,5,19,21.
Nivelurile ridicate de arginină plasmatică deosebesc în cele din urmă ARG1-D de alte UCD, precum și de tulburările neurometabolice și neurologice, cum ar fi paralizia cerebrală sau paraplegia spastică ereditară2,4,12.
Referințe:
1. Diez-Fernandez C, et al. Hum Mutat. 2018;39:1029-1050. 2. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 3. Häberle J, et al. J Inherit Metab Dis. 2019;1–39. 4. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 5. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 6. Sun A, et al. Arginase deficiency. In: Adams MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 7. Diaz GA, et al. Poster presented at: 13th European Paediatric Neurology Society (EPNS) Congress; September 17-21, 2019; Athens, Greece. Poster P06-34. 8. Carvalho DR, et al. Gene. 2012;509:124-130. 9. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 10. Barmore W, et al. Physiology, Urea Cycle. [Updated 2021 May 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan. Available at: https://www.ncbi.nlm.nih.gov/books/NBK513323. Accessed December 3, 2021. 11. Tapiero H, et al. Biomed Pharmacother. 2002;56:439-445. 12. Prasad A, et al. J Child Neurol. 1997;12:301-309. 13. Amayreh W, et al. Dev Med Child Neurol. 2014;56:1021-1024. 14. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 15. NORD. The Physician’s Guide to Urea Cycle Disorders. 2012. Available at: http://www.nucdf.org/documents/NORD_Physician_Guide_to_Urea_Cycle_Disorders.pdf. Accessed November 26, 2021. 16. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 17. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 18. Sin YY, et al. J Mol Med (Berl). 2015;93:1287-1296. 19. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 20. Bakhiet M, et al. Medicine (Baltimore). 2018;97:e10780. 21. Schlune A, et al. Amino Acids. 2015;47:1751-1762.