Manifestările ARG1-D le imită adesea pe cele ale altor tulburări neurologice și neurometabolice, cum ar fi alte tulburări ale ciclului ureei (UCD), paralizia cerebrală (PC) sau paraplegia spastică ereditară (HSP)5,6.
Diagnosticul diferențial al ARG1-D implică identificarea manifestărilor clinice asociate cu nivelurile ridicate de arginină plasmatică4-7.
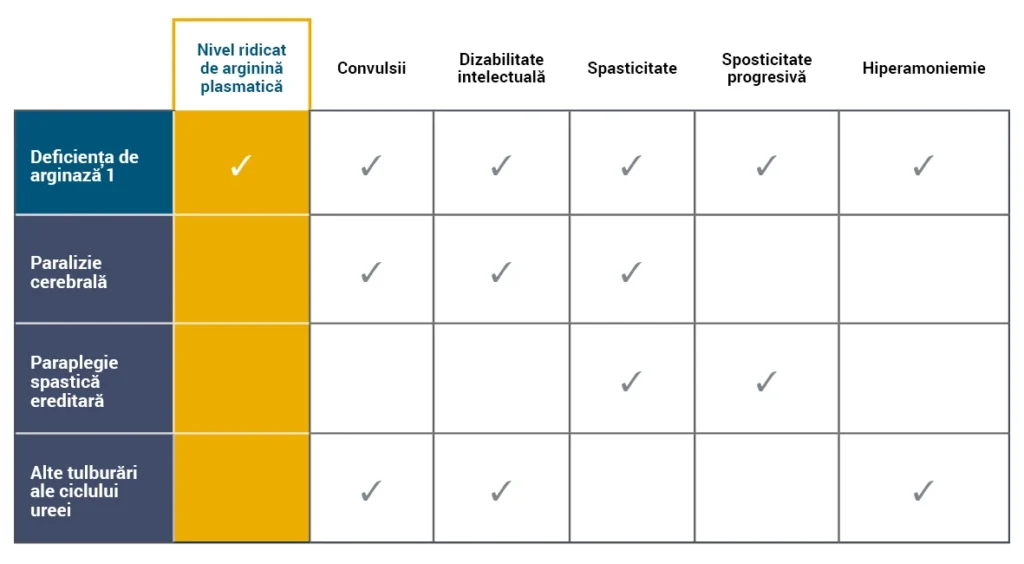
- Hiperamoniemia nu este un semn distinctiv al ARG1-D și episoadele acute de hiperamoniemie apar rar4,8.
Din cauza limitărilor screening-ului nou-născutului, ARG1-D poate fi omis din numeroase motive:
- Determinarea nivelurilor limită de arginină în screening este problematică, deoarece transferul metaboliților, cum ar fi arginina, de la mamă la copil poate compromite testarea9,10.
- Algoritmii de screening și nivelurile limită de arginină variază9.
- ARG1-D nu este inclus în panourile de screening pentru nou-născuți în majoritatea țărilor europene11.
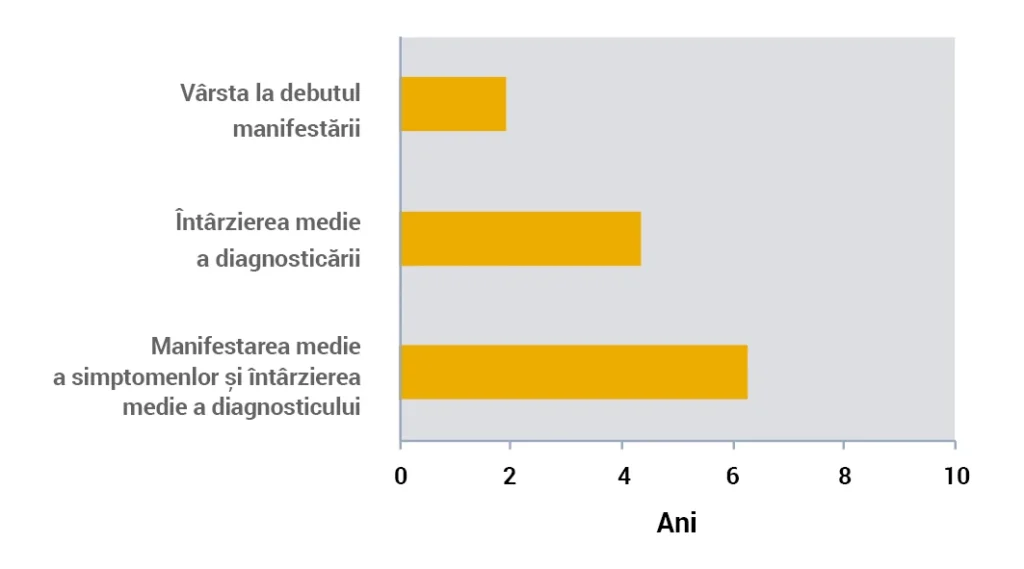
Întârzierile în diagnosticare, împreună cu debutul tardiv al simptomelor, duc la intervenția inițială numai la vârsta de ~6 ani1.
Testarea de rutină cu un grupului de aminoacizi din plasmă urmată de un test genetic poate confirma ARG1-D12,13.
Înainte de inițierea diagnosticului, este important să se evalueze complet istoricul medical, alimentar, familial și social și să se efectueze un examen fizic amănunțit.
Verificarea nivelurilor ridicate de arginină, care provoacă manifestările ARG1-D, cu teste de rutină3,12,13.


Dacă sunt prezente niveluri ridicate de arginină plasmatică, un test genetic† poate confirma diagnosticul.
†Datorită heterogenității genetice a genotipurilor ARG1, nu au fost identificate toate mutațiile care cauzează ARG1-D.
Referințe:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.